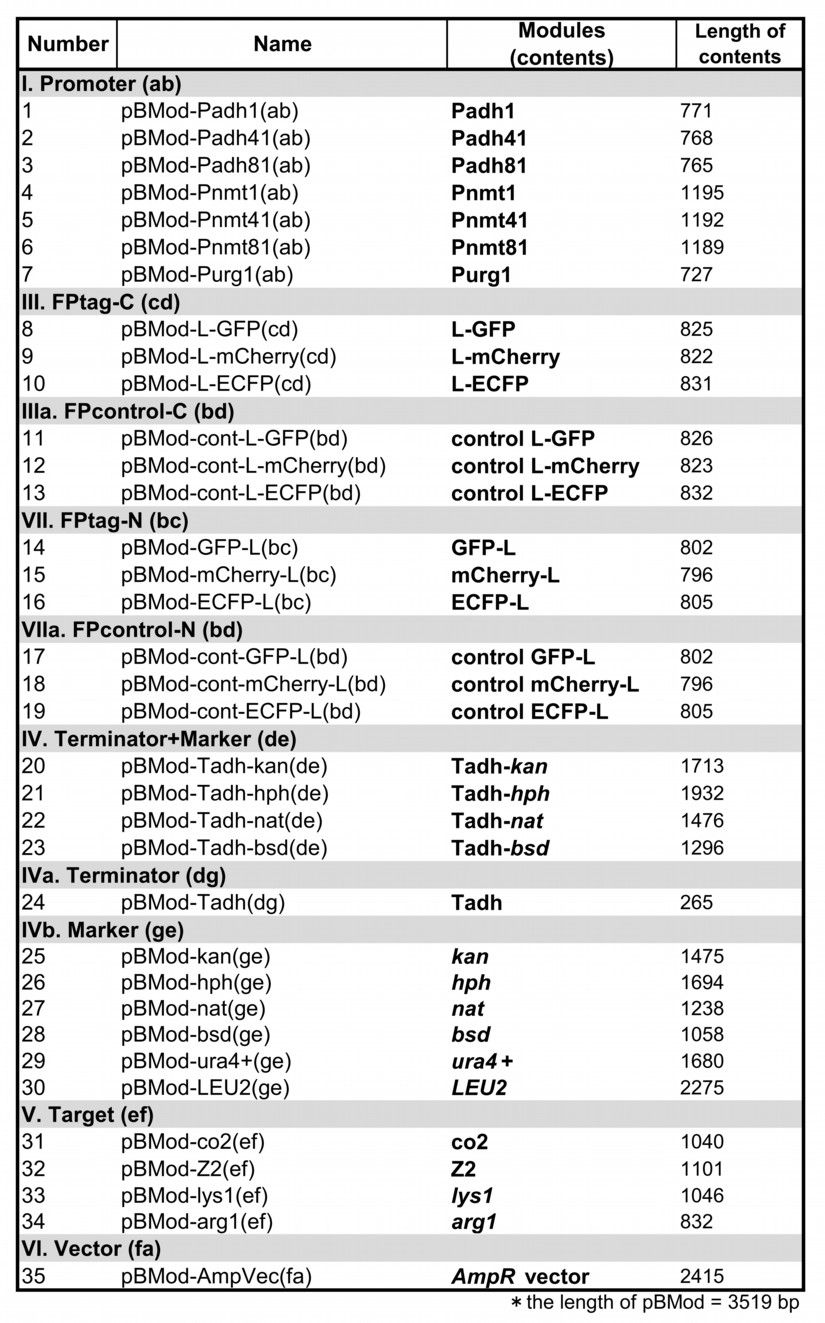
1-1.使用するパーツ(module)を手元に揃える。
▶パーツプラスミド(module plasmid)のライブラリーから必要なものを選ぶ。
▶発現させたい遺伝子ORFのPCR増幅断片を用意する。テンプレートの混入を避けるために切り出し精製を行う。
1-2.使用するパーツ(module)の濃度を揃える。
▶各module plasmidsを25倍希釈し、濃度(ng / μl)を吸光度計を用いて測定する。
▶各module plasmidsをddH2Oにて 20 fmol / μlに希釈する。加えるddH2Oの量は下記の計算式に則って計算する。
*吸光度計による測定結果をA (ng / μl), module plasmidの長さを B (bp)とする。
各module plasmidの長さはこのページの下部に掲載した。
module plasmid 4 μlに対し加えるddH2Oの量 (μl) = ![]()
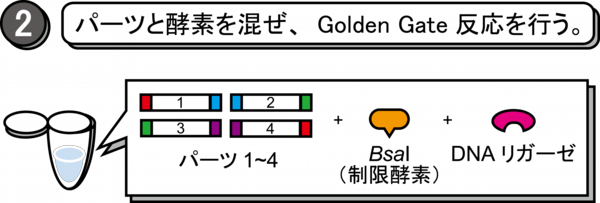
2-1.以下の組成でPCRチューブ内に反応液を調製する。
BsaI (NEB, #R0535S) 1.5 μl
T4 DNA ligase (Promega, #M1794) 0.5 μl
10x T4 DNA ligase buffer 2 μl
module plasmids (20 fmol / μl) 各1 μl
ddH2O up to 20 μl
total 20 μl
*T4 DNA ligase bufferは析出しやすいので、ボルテックスにてよく懸濁し使用する。
2-2.以下のサイクルでGolden Gate反応を行う。
1: 37℃ 1 min
2: 37℃ 2 min
3: 16℃ 5 min
4: go to step 2 additional 49 times
5: 50℃ 10 min
6: 80℃ 10 min
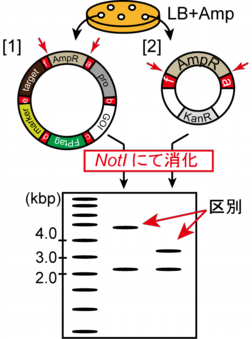
3-1.Golden Gate反応終了後、反応液 2 μlを大腸菌 100 μlに加え、形質転換を行う。LB+Ampプレートにまく。
3-2.生えてきた大腸菌コロニーを液体培養し、プラスミドを抽出する。
3-3.抽出したプラスミドを制限酵素NotI にて消化し、確認を行う。
▶LB+Ampプレートによる選択のみでは、Golden Gate法によりligationされたplasmid [1]とVector moduleそのもの [2] を区別することができないため、抽出したプラスミドをNotI で消化し、電気泳動を行う。
▶これによりVector moduleにクローニングされているDNAの断片長が分かるので、両者([1]と[2])を区別することができる。
▶ligationに成功したプラスミドを制限酵素FseIにより消化し線状化することで、分裂酵母染色体へのintegrationが可能となる。
▶FseIによる消化がうまくいかない場合は、プラスミドをフェノール・クロロホルム抽出あるいはカラムにて精製する。
▶integration後の選択は、kan, hph などの選択マーカーを利用して行う。
[新moduleを作製する際の留意点]
・全てのmoduleの末端の配列は 5'-tttGGTCTCaNNNN-3' である('N'はmoduleの種類により異なる)。
・cohesive end 'b' (=TATG) は翻訳のための開始コドンを含んでいる。
・cohesive end 'c' (=CTG A) によって読み枠がずれるのを防ぐために、'c'の3'側に適当な2塩基を追加し次のmoduleを繋げる。
・cohesive end 'd' (=GTCT) の5'側には翻訳を停止させるための終止コドンを入れる。
・Vector (fa) module の両端にはNotI の認識サイトを付加した上でBsaIのサイトをつける。
(5'-tttGGTCTCaGCGGaaGCGGCCGC-(specific 20b)-3')
・作製されたプラスミドがBsaIによって30分~90分で消化可能であることを確認する。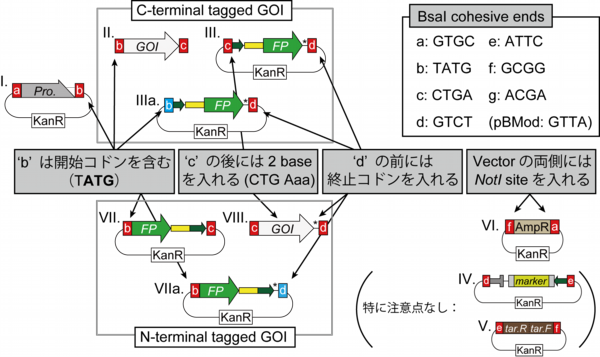
[補足]
各moduleの長さ